Stacks is a software pipeline for building loci from short-read sequences, such as those generated on
the Illumina platform. Stacks was developed to work with restriction enzyme-based data, such as RAD-seq, for the
purpose of building genetic maps and conducting population genomics and phylogeography.
Stacks Pipeline

Genetic Maps
Stacks can be used to generate mappable markers from RAD-seq data. Thousands of markers
can be generated from a single generation, F1 map as well as markers for traditional F2 and
backcross designs. Stacks can export data to JoinMap, OneMap, or R/qtl. These data can be used for
examining genomic structure as well as assembling genomic assemblies.
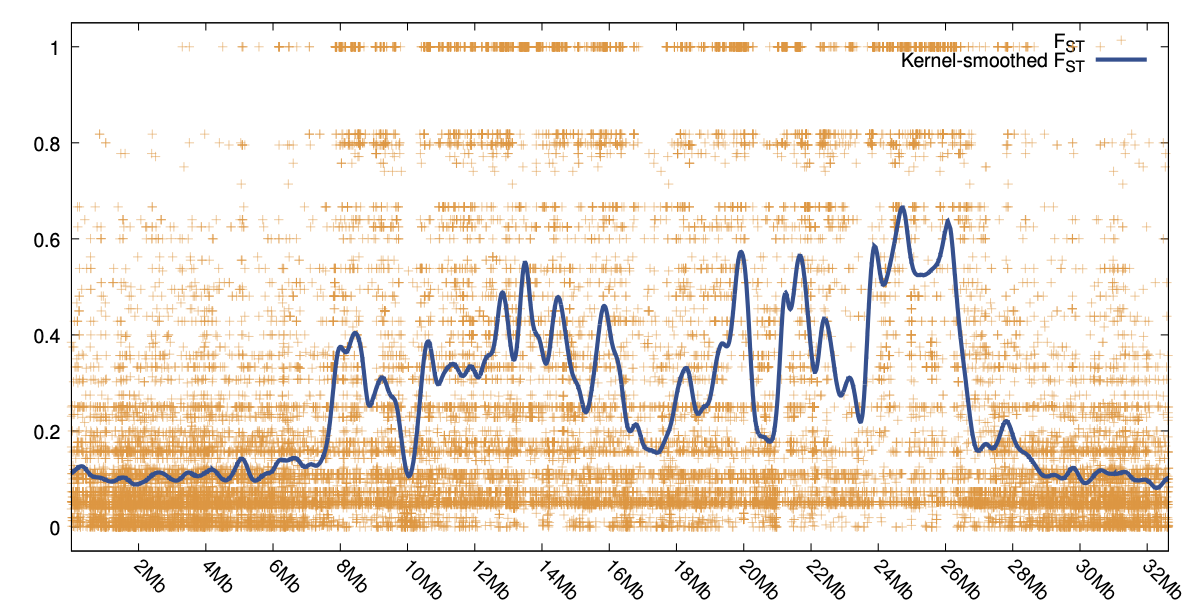
Population Genomics
Stacks can be used to identify SNPs within or among populations. Stacks provides tools to generate
summary statistics and to compute population genetic measures such as FIS and π within populations
and FST between populations, allowing for genome scans. Data can be exported in
VCF format and for use in programs such as
STRUCTURE or GenePop.
Data can also be exported for cline analysis in HZAR format.
Any SNP dataset in VCF format can also be imported into the Stacks
populations module. SNPs generated from re-sequencing or RNA-seq, among other methods, can now be filtered/smoothed
in the same way RAD data can.
Phylogenetics
Stacks can export GBS/RAD data for phylogenetic analysis. Identified SNPs can be concatenated and exported in
Phylip format; these SNPs can be specified
as fixed within and variable among populations, or simply all variable sites (encoded in
IUPAC notation). Stacks can also export SNPs with their full flanking sequence -- the RAD locus. These data can be exported in Phylip
format (either as concatenated or partitioned data) which can be fed into any standard phylogenetics package
such as PhyML or RAxML.
|
Getting started with Stacks
Frequently Asked Questions
|
The Stacks pipeline is designed modularly to perform several different
types of analyses. Programs listed under Raw Reads are used
to clean and filter raw sequence data. Programs under Core
represent the main Stacks pipeline — building loci (ustacks),
creating a catalog of loci (cstacks, and matching samples back against
the catalog (sstacks), transposing the data (tsv2bam), adding
paired-end reads to the analysis and calling genotypes, and population
genomics analysis. Programs under Execution Control will run
the whole pipeline.
Raw reads
|
Core
|
Execution control
|
Utility programs
|
The process_radtags program examines raw reads from an Illumina sequencing run and
first, checks that the barcode and the restriction enzyme cutsite are intact (correcting minor errors).
Second, it slides a window down the length of the read and checks the average quality score within the window.
If the score drops below 90% probability of being correct, the read is discarded. Reads that pass quality
thresholds are demultiplexed if barcodes are supplied.
The process_shortreads program performs the same task as process_radtags
for fast cleaning of randomly sheared genomic or transcriptomic data. This program will trim reads that are below the
quality threshold instead of discarding them, making it useful for genomic assembly or other analyses.
The clone_filter program will take a set of reads and reduce them according to PCR
clones. This is done by matching raw sequence or by referencing a set of random oligos that have been included in the sequence.
The kmer_filter program allows paired or single-end reads to be filtered according to the
number or rare or abundant kmers they contain. Useful for both RAD datasets as well as randomly sheared genomic or
transcriptomic data.
The ustacks program will take as input a set of short-read sequences and align them into
exactly-matching stacks. Comparing the stacks it will form a set of loci and detect SNPs at each locus using a
maximum likelihood framework.
A catalog can be built from any set of samples processed
by the ustacks program. It will create a set of consensus loci, merging alleles together. In the case
of a genetic cross, a catalog would be constructed from the parents of the cross to create a set of
all possible alleles expected in the progeny of the cross.
Sets of stacks constructed by the ustacks
program can be searched against a catalog produced by the cstacks program. In the case of a
genetic map, stacks from the progeny would be matched against the catalog to determine which progeny
contain which parental alleles.
The tsv2bam program will transpose data so that it is oriented by locus, instead of by sample.
In additon, if paired-ends are available, the program will pull in the set of paired reads that are associate with each
single-end locus that was assembled de novo.
The gstacks - For de novo analyses, this program will pull in paired-end
reads, if available, assemble the paired-end contig and merge it with the single-end locus, align reads
to the locus, and call SNPs. For reference-aligned analyses, this program will build loci from the
single and paired-end reads that have been aligned and sorted.
This populations program will compute population-level summary statistics such
as π, FIS, and FST. It can output site level SNP calls in VCF format and
can also output SNPs for analysis in STRUCTURE or in Phylip format for phylogenetics analysis.
The denovo_map.pl program executes each of the Stacks components to create a genetic
linkage map, or to identify the alleles in a set of populations.
The ref_map.pl program takes reference-aligned input data and executes each of the Stacks
components, using the reference alignment to form stacks, and identifies alleles. It can be used in a genetic map
of a set of populations.
The load_radtags.pl program takes a set of data produced by either the denovo_map.pl or
ref_map.pl progams (or produced by hand) and loads it into the database. This allows the data to be generated on
one computer, but loaded from another. Or, for a database to be regenerated without re-executing the pipeline.
The stacks-dist-extract script will pull data distributions from the log and distribs
files produced by the Stacks component programs.
The stacks-integrate-alignments script will take loci produced by the de novo pipeline,
align them against a reference genome, and inject the alignment coordinates back into the de novo-produced data.
The stacks-private-alleles script will extract private allele data from the populations program
outputs and output useful summaries and prepare it for plotting.
