populations
The populations program will analyze a population of individual samples computing a number
of population genetics statistics as well as exporting a variety of standard output formats. A population map
specifying which individuals belong to which population is submitted to the program and the program will then
calculate population genetics statistics such as expected/observed heterozygosity, π, and FIS at each
nucleotide position. The populations program will compare all populations pairwise to
compute FST. If a set of data is reference aligned, then a kernel-smoothed FST will also be
calculated. The populations program can also compute a number of haplotype-based
population genetics statistics including haplotype diversity, ΦST, and FST’. For
more information on how to specify a population map, see the manual.
The populations program provides strong filtering options to only include loci or variant sites that occur at
certain frequencies in each population or in the metapopulation. In addition, the program accepts
whitelists and blacklists if you want to include a specific list of loci (or exclude a specific
list of loci). For more information on whitelists and blacklists, see the manual.
Program Options
populations -P dir [-O dir] [-M popmap] (filters) [--fstats] [-k [--sigma=150000] [--bootstrap [-N 100]]] (output formats)
populations -V vcf -O dir [-M popmap] (filters) [--fstats] [-k [--sigma=150000] [--bootstrap [-N 100]]] (output formats)
- -P,--in_path — path to the directory containing the Stacks files.
- -V,--in_vcf — path to an input VCF file.
- -O,--out_path — path to a directory where to white the output files. (Required by -V; otherwise defaults to value of -P.)
- -M,--popmap — path to a population map. (Format is 'SAMPLE1 \t POP1 \n SAMPLE2 ...'.)
- -t,--threads — number of threads to run in parallel sections of code.
Data Filtering:
- -p,--min-populations [int] — minimum number of populations a locus must be present in to process a locus.
- -r,--min-samples-per-pop [float] — minimum percentage of individuals in a population required to process a locus for that population.
- -R,--min-samples-overall [float] — minimum percentage of individuals across populations required to process a locus.
- -H,--filter-haplotype-wise — apply the above filters haplotype wise (unshared SNPs will be pruned to reduce haplotype-wise missing data).
- --min-maf [float] — specify a minimum minor allele frequency required to process a nucleotide site at a locus (0 < min_maf < 0.5; applied to the metapopulation).
- --min-mac [int] — specify a minimum minor allele count required to process a SNP (applied to the metapopulation).
- --max-obs-het [float] — specify a maximum observed heterozygosity required to process a nucleotide site at a locus (applied to the metapopulation).
- --min-gt-depth [int] — specify a minimum number of reads to include a called SNP (otherwise marked as missing data).
- --write-single-snp — restrict data analysis to only the first SNP per locus.
- --write-random-snp — restrict data analysis to one random SNP per locus.
- -B,--blacklist — path to a file containing Blacklisted markers to be excluded from the export.
- -W,--whitelist — path to a file containing Whitelisted markers to include in the export.
Locus stats:
- --hwe — calculate divergence from Hardy-Weinberg equilibrium for each locus.
Fstats:
- --fstats — enable SNP and haplotype-based F statistics.
- --fst_correction — specify a correction to be applied to Fst values: 'p_value'. Default: off.
- --p_value_cutoff [float] — maximum p-value to keep an Fst measurement. Default: 0.05.
Kernel-smoothing algorithm:
- -k,--smooth — enable kernel-smoothed π, FIS, FST, FST', and ΦST calculations.
- --smooth-fstats — enable kernel-smoothed FST, FST', and ΦST calculations.
- --smooth-popstats — enable kernel-smoothed π and FIS calculations. (Note: turning on smoothing implies --ordered_export.)
- --sigma [int] — standard deviation of the kernel smoothing weight distribution. Sliding window size is defined as 3xσ, default σ = 150kbp (3xσ = 450kbp).
- --bootstrap-archive — archive statistical values for use in bootstrap resampling in a subsequent run, statistics must be enabled to be archived.
- --bootstrap — turn on boostrap resampling for all kernel-smoothed statistics.
- -N,--bootstrap-reps [int] — number of bootstrap resamplings to calculate (default 100).
- --bootstrap-pifis — turn on boostrap resampling for smoothed SNP-based π and FIS calculations.
- --bootstrap-fst — turn on boostrap resampling for smoothed FST calculations based on pairwise population comparison of SNPs.
- --bootstrap-div — turn on boostrap resampling for smoothed haplotype diveristy and gene diversity calculations based on haplotypes.
- --bootstrap-phist — turn on boostrap resampling for smoothed φST calculations based on haplotypes.
- --bootstrap-wl [path] — only bootstrap loci contained in this whitelist.
File output options:
- --ordered-export — if data is reference aligned, exports will be ordered; only a single representative of each overlapping site.
- --fasta-loci — output locus consensus sequences in FASTA format..
- --fasta-samples — output the sequences of the two haplotypes of each (diploid) sample, for each locus, in FASTA format.
- --vcf — output SNPs and haplotypes in Variant Call Format (VCF).
- --vcf-all — output all sites in Variant Call Format (VCF).
- --genepop — output results in GenePop format.
- --structure — output results in Structure format.
- --radpainter — output results in fineRADstructure/RADpainter format.
- --plink — output genotypes in PLINK format.
- --hzar — output genotypes in Hybrid Zone Analysis using R (HZAR) format.
- --phylip — output nucleotides that are fixed-within, and variant among populations in Phylip format for phylogenetic tree construction.
- --phylip-var — include variable sites in the phylip output encoded using IUPAC notation.
- --phylip-var-all — include all sequence as well as variable sites in the phylip output encoded using IUPAC notation.
- --treemix — output SNPs in a format useable for the TreeMix program (Pickrell and Pritchard).
- --no-hap-exports — omit haplotype outputs.
- --fasta-samples-raw — output all haplotypes observed in each sample, for each locus, in FASTA format.
Genetic map output options (population map must specify a genetic cross):
- --map-type — genetic map type to write. 'CP', 'DH', 'F2', and 'BC1' are the currently supported map types.
- --map-format — mapping program format to write, 'joinmap', 'onemap', and 'rqtl' are currently supported.
Additional options:
- -h,--help— display this help messsage.
- -v,--version— print program version.
- --verbose— turn on additional logging.
- --log-fst-comp— log components of FST/ΦST calculations to a file.
Kernel-smoothing statistical values along a reference genome
If your data are aligned to a reference genome, then it is possible to ask populations to produce
a kernel-smoothed average of your statistical point values along the genome. This translates your point values into a continuous
set of values along a chromosome/scaffold. If --smooth is enabled, for each per-population statistical
value, including FIS, π, and the haplotype-level measures of π; and for each population-pair value, such as
FST, and the haplotype-level values of DXY, φST, etc. will be smoothed along the chromosome.
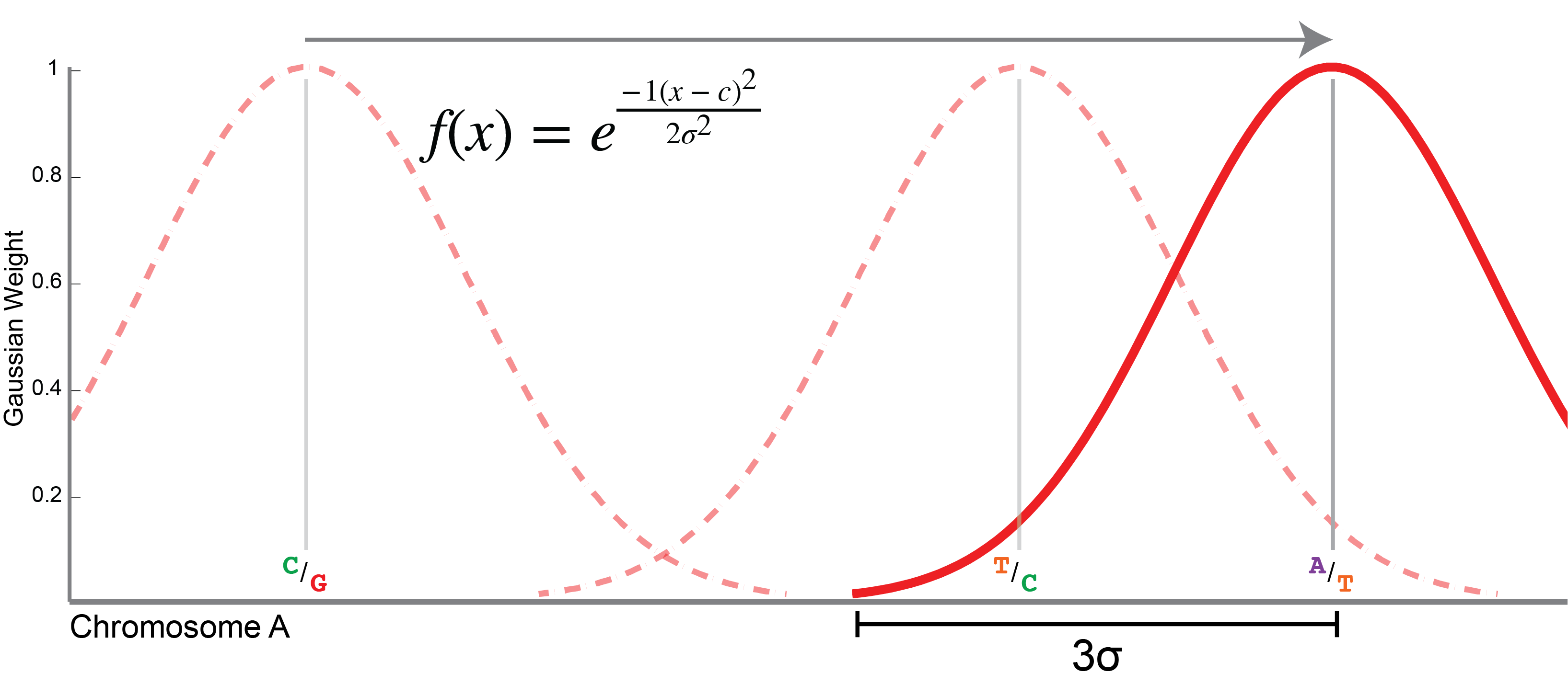
This is diagrammed in the image to the right. The sliding window is defined by a Gaussian distribution (pictured as a red arc,
with f(x) defined in the image). The window is centered over each successive variant site along the chromosome. The size
of the window is determined by σ or one standard deviation of the Gaussian distribution. The width of the window, in each
direction, then, is 3 x σ, or three standard deviations in length (marked in the figure). The sigma value is by
default 150 Kbp in length, giving a window that is 3 x 150 = 450 Kbp on one side, or 900 Kbp in total length (including both
tails). This value can be set using the --sigma option to populations.
The smoothed values are calculated by weighting the central value (the variant site the window is centered on), by all of the nearby
values — stretching out 3σ in either direction. These nearby values are weighted more heavily when they are closer to the
central value. The Gaussian distribution naturally gives the weights for the nearby values.
So, increasing σ (with the --sigma option) will smooth values out further and decreasing it will give
a more coarse, or jagged set of values. If your genome is large, it makes sense to increase the size of the window and vice-versa.
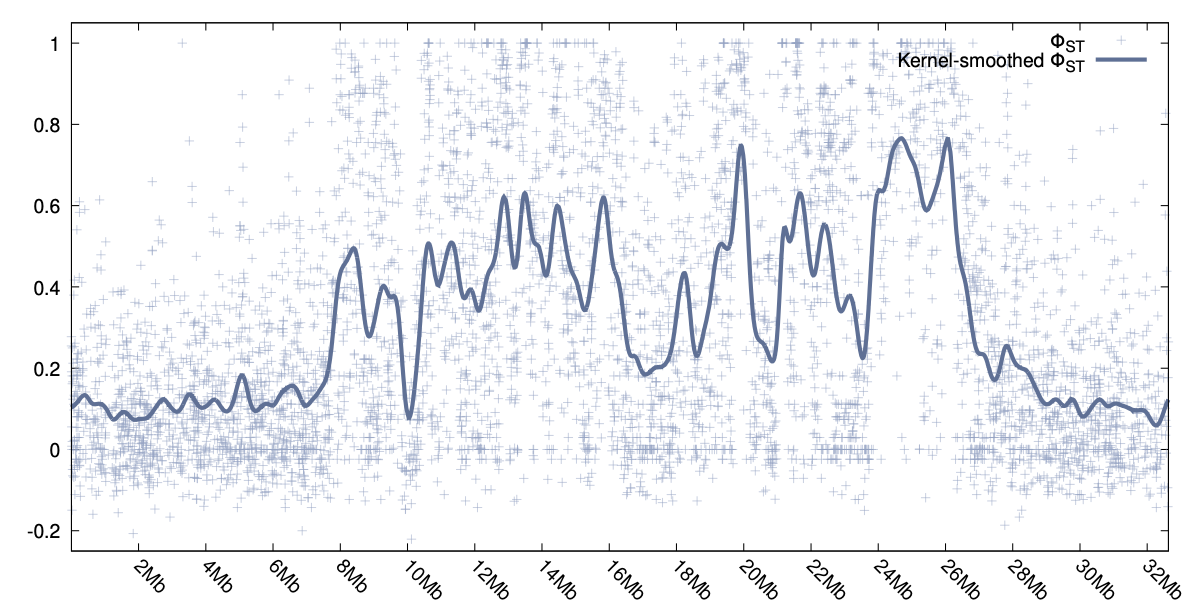
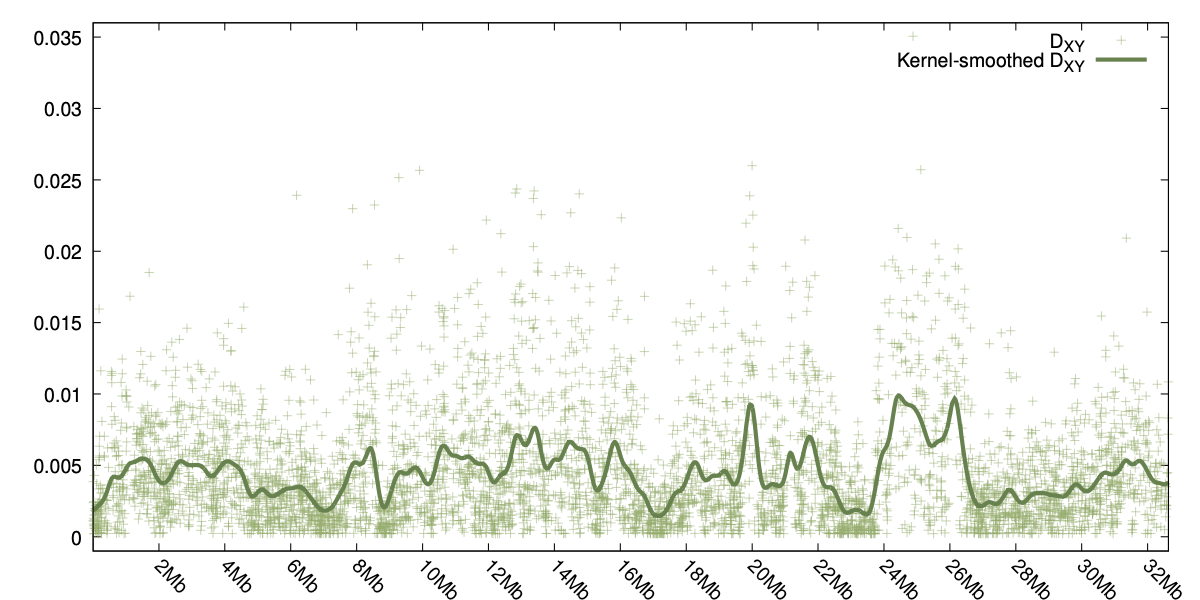
Two examples can be seen to the right, one for φST and the other showing the smoothed values for DXY.
Bootstrap resampling
The bootstrap resampling procedure is designed to determine the statistical significance of a particular sliding window
value relative to the generated (bootstrapped) empirical distribution. Bootstrap resampling will generate a p-value describing the
statistical significance of a particular kernel-smoothed sliding window. Bootstrap resampling allows us to move from single point
values being statistically significant (or not), to regions of the genome being statistically significant (or not).
In Stacks v2, bootstrapping is a two stage process that requires the data be aligned to a reference genome. First, the population
statistical values must be archived, and then in a subsequent run, this archive of values can be pre-loaded so it is available as
a source for the resampling proceedure.
The bootstrap resampling process will center a window on each variable nucleotide position in the population and resample it X
times (with replacement), and then calculate a p-value. Bootstrap resampling can be applied to all smoothed values, including
the population summary statistics FIS, Π, and haplotype diversity, as well as the calculation of FST
and ΦST between pairs of populations. If you have tens of thousands of variable sites (not unusual) and lots
of populations, this calculation has to be repeated for every variable site in each population to bootstrap the summary
statistics and for all variable sites between each pair of populations for FST and ΦST. So, bootstrap
resampling can take a while. The run time can be decreased by enabling multiple threads (--threads) to the
populations program.
In Stacks v2, the populations program only loads one chromosome/scaffold at a time into memory. So, when
computing smoothed population statistics on chromosome 1, populations has not loaded or computed the
corresponding values on chromosome 2, 3, etc. To conduct bootstrapping for sliding windows on chromosome 1, populations
must be able to resample from the full set of statistical values (that have not yet been computed). To maintain efficency, bootstrapping
is done over two or more runs. During the first run, populations is instructed to archive statistical values
with the --bootstrap-archive flag. This flag will cause the file, populations.bsarchive.tsv
to be created and populated in the populations output directory. This file contains all the population statistics
stored in a very simple and efficient text file. If a particular statistic is not enabled during this
stage (e.g. --fstats), than those statistical values will not be available for bootstrap resampling.
Once the archive is created, the user can re-run populations, selecting one of the standard --bootstrap*
flags to enable resampling. This can be done as many times as is desired, as long as the archive remains.
Since bootstrapping is so computationally intensive, there are several command line options to the
populations program to allow one to turn bootstrapping on for only a subset of the statistics. In
addition, a bootstrap "whitelist" is available so you can choose to only bootstrap certain loci (say the loci on a single
chromosome). This allows one to take the following strategy for bootstrapping to appropriate levels:
- Bootstrap all loci (for example) to 1,000 repetitions.
- Identify those loci that are below some p-value threshold (say 0.05).
- Add these loci to the bootstrapping whitelist.
- Bootstrap again to 10,000 repetitions (now only those loci in the whitelist will be bootstrapped).
- Identify those loci that are below some p-value threshold (say 0.005)
- Add these loci to the bootstrapping whitelist.
- Bootstrap again to 100,000 repetitions (now only those loci in the whitelist will be bootstrapped).
- And so on to the desired level of significance...
If instead you are interested in the statistical significance of a particular point estimate of an FST
measure, you will want to use the p-value from Fisher's Exact Test, which is calculated for each variable position
between pairs of populations and is provided in the FST output files.
Example Usage
- Calculate population statistics in a single population and output a Variant Call Format (VCF) SNP file. Run populations on 8 processors:
~/% populations -P ./stacks/ --vcf -t 8
- Include multiple populations using a population map, and turn on kernel smoothing for π, FIS, and FST (data must be reference aligned for smoothing):
~/% populations -P ./stacks/ --popmap ./samples/popmap --smooth -t 8
- Filter input so that to process a locus it must be present in 10 of the populations and in 75% of individuals in each population:
~/% populations -P ./stacks/ --popmap ./samples/popmap --smooth -p 10 -r 0.75 -t 8
- Output data for STRUCTURE and in the GenePop format. Only write the first SNP from any RAD
locus, to prevent linked data from being processed by STRUCTURE:
~/% populations -P ./stacks/ --popmap ./samples/popmap --smooth -p 10 -r 0.75 -f p_value -t 8 --structure --genepop --write-single-snp
- Include a whitelist of 1,000 random loci so that we output a computationally manageable amount of data to STRUCTURE:
~/% populations -P ./stacks/ --popmap ./samples/popmap --smooth -p 10 -r 0.75 -f p_value -t 12 --structure --genepop --write-single-snp -W ./wl_1000
Here is one method to generate a list of random loci from a populations summary statistics file (this command goes all on one line):
~/% grep -v "^#" populations.sumstats.tsv |
cut -f 1 |
sort |
uniq |
shuf |
head -n 1000 |
sort -n > whitelist.tsv
This command does the following at each step:
- Grep pulls out all the lines in the sumstats file, minus the commented header lines. The sumstats file contains all the polymorphic loci in the analysis.
- cut out the second column, which contains locus IDs
- sort those IDs
- reduce them to a unique list of IDs (remove duplicate entries)
- randomly shuffle those lines
- take the first 1000 of the randomly shuffled lines
- sort them again and capture them into a file.
- Invoke bootstrap resampling to generate p-values reflecting the statistical significance of regions of the genome defined by kernel-smoothed statistics. First,
we have to run populations and instruct it to archive our statistical values so they are available for the bootstrapping process:
~/% populations -P ./stacks --popmap ../popmap.tsv --fstats --bootstrap-archive -t 14
Second, once we have generated the archive, we can re-execute populations and tell it to bootstrap resample. Here we also increase
the default number of repetitions to 1000. We could also choose one of the --bootstrap-* flags to only bootstrap a particular
type of statistic we are interested in to save time:
~/% populations -P ./stacks --popmap ../popmap.tsv --fstats --bootstrap --bootstrap-reps 1000 -t 14
Program Outputs
The populations program has a number of standard outputs, besides any particular export that is asked for. The formats of
these files are described in detail in the manual.
- populations.log: This file contains everything output on the screen, including the version number of Stacks and when
it was run. It also includes a lot of useful information describing the data after filters have been applied, including how many loci were
discarded/kept, the mean length of loci, and mean number of sites.
- populations.log.distribs: This file contains a number of distributions describing the data. The distribution are
provided in two forms: prior to filtering and after filtering. You can view the file manually, or you can use the
stacks-dist-extract utility we provide to pull out a particular distribution. One distribution that can tell you
a lot about your data is the number of samples per locus, pre- and post-filtering. You can extract it like this:
% stacks-dist-extract populations.log.distribs samples_per_loc_prefilters
# Distribution of valid samples matched to a catalog locus prior to filtering.
n_samples n_loci
1 17857
2 6103
3 3855
4 4253
5 5591
6 2786
7 3264
8 6518
9 41362
10 40333
You can see in this data set that more than 40k loci each were found in 9 or 10 samples. But that 17k loci were only found in a single sample -- these loci are likely
unreliable.
When we look at this distribution after filtering, we can see the effect of filtering out the low-value loci:
% stacks-dist-extract populations.log.distribs samples_per_loc_postfilters
# Distribution of valid samples matched to a catalog locus after filtering.
n_samples n_loci
8 3040
9 41362
10 40333
- populations.sumstats.tsv: This file contains summary statistics describing every SNP found in every population defined in the
population map. This includes the frequecy of alleles, expected/observed heterozygosity, π, FIS, and so on.
- populations.hapstats.tsv: This file contains summary statistics describing every locus (phased SNPs) found in every population defined in the
population map. This includes the frequecy of haplotypes, gene and haplotype diversity.
- populations.sumstats_summary.tsv: This file contains population-level, mean summary statistics.
- If F-statistics was enabled, populations.fst_X-Y.tsv: This file contains SNP-level measures of FST.
- If F-statistics was enabled, populations.phistats_X-Y.tsv: This file contains haplotype-level measures of FST.
Other Pipeline Programs
Raw reads
|
Core
|
Execution control
|
Utility programs
|
The process_radtags program examines raw reads from an Illumina sequencing run and
first, checks that the barcode and the restriction enzyme cutsite are intact (correcting minor errors).
Second, it slides a window down the length of the read and checks the average quality score within the window.
If the score drops below 90% probability of being correct, the read is discarded. Reads that pass quality
thresholds are demultiplexed if barcodes are supplied.
The process_shortreads program performs the same task as process_radtags
for fast cleaning of randomly sheared genomic or transcriptomic data. This program will trim reads that are below the
quality threshold instead of discarding them, making it useful for genomic assembly or other analyses.
The clone_filter program will take a set of reads and reduce them according to PCR
clones. This is done by matching raw sequence or by referencing a set of random oligos that have been included in the sequence.
The kmer_filter program allows paired or single-end reads to be filtered according to the
number or rare or abundant kmers they contain. Useful for both RAD datasets as well as randomly sheared genomic or
transcriptomic data.
The ustacks program will take as input a set of short-read sequences and align them into
exactly-matching stacks. Comparing the stacks it will form a set of loci and detect SNPs at each locus using a
maximum likelihood framework.
A catalog can be built from any set of samples processed
by the ustacks program. It will create a set of consensus loci, merging alleles together. In the case
of a genetic cross, a catalog would be constructed from the parents of the cross to create a set of
all possible alleles expected in the progeny of the cross.
Sets of stacks constructed by the ustacks
program can be searched against a catalog produced by the cstacks program. In the case of a
genetic map, stacks from the progeny would be matched against the catalog to determine which progeny
contain which parental alleles.
The tsv2bam program will transpose data so that it is oriented by locus, instead of by sample.
In additon, if paired-ends are available, the program will pull in the set of paired reads that are associate with each
single-end locus that was assembled de novo.
The gstacks - For de novo analyses, this program will pull in paired-end
reads, if available, assemble the paired-end contig and merge it with the single-end locus, align reads
to the locus, and call SNPs. For reference-aligned analyses, this program will build loci from the
single and paired-end reads that have been aligned and sorted.
This populations program will compute population-level summary statistics such
as π, FIS, and FST. It can output site level SNP calls in VCF format and
can also output SNPs for analysis in STRUCTURE or in Phylip format for phylogenetics analysis.
The denovo_map.pl program executes each of the Stacks components to create a genetic
linkage map, or to identify the alleles in a set of populations.
The ref_map.pl program takes reference-aligned input data and executes each of the Stacks
components, using the reference alignment to form stacks, and identifies alleles. It can be used in a genetic map
of a set of populations.
The load_radtags.pl program takes a set of data produced by either the denovo_map.pl or
ref_map.pl progams (or produced by hand) and loads it into the database. This allows the data to be generated on
one computer, but loaded from another. Or, for a database to be regenerated without re-executing the pipeline.
The stacks-dist-extract script will pull data distributions from the log and distribs
files produced by the Stacks component programs.
The stacks-integrate-alignments script will take loci produced by the de novo pipeline,
align them against a reference genome, and inject the alignment coordinates back into the de novo-produced data.
The stacks-private-alleles script will extract private allele data from the populations program
outputs and output useful summaries and prepare it for plotting.