This tutorial will describe how three major Stacks parameters function to control the de novo assembly of loci by the ustacks and cstacks programs. All three of these parameters can be specified to the pipeline wrapper, denovo_map.pl, which will then pass them through to the appropriate pipeline component. Or, if you are running the pipeline components by hand, two of them are specified to ustacks (and are applied to each sample processed by Stacks) and one of them to cstacks, which builds the catalog of loci for the population of samples. The following table summarizes the three parameters:
| Parameter Description | denovo_map.pl Parameter | Pipeline component | Component Parameter | Default Value |
|---|---|---|---|---|
| Minimum stack depth / minimum depth of coverage | -m | ustacks | -m | 3 |
| Distance allowed between stacks | -M | ustacks | -M | 2 |
| Distance allowed between catalog loci | -n | cstacks | -n | 1 |
To jump ahead to a particular parameter click below:

Figure 1. The initial stage of the ustacks de novo assembly algorithm forms exactly matching stacks from raw short-reads.
The ustacks program is executed on each sample in a population (each of the parents and progeny in a mapping cross, or each individual sample or pooled sample in a population analysis) and loci from that sample are reconstructed. This is done in two major steps, first, exactly matching reads are stacked together out of the raw data. These stacks can be thought of generally as representing alleles, although some of them will represent errors.
The minimum stack depth parameter controls the number of raw reads required to form an initial stack. If the depth of coverage for a particular stack is below this value, then an allele will not be formed and those reads are temporarily set aside by the algorithm (they are used later in the algorithm, see below). Raw reads that are placed in a stack are refered to as primary reads, while those that are set aside are referred to as secondary reads.
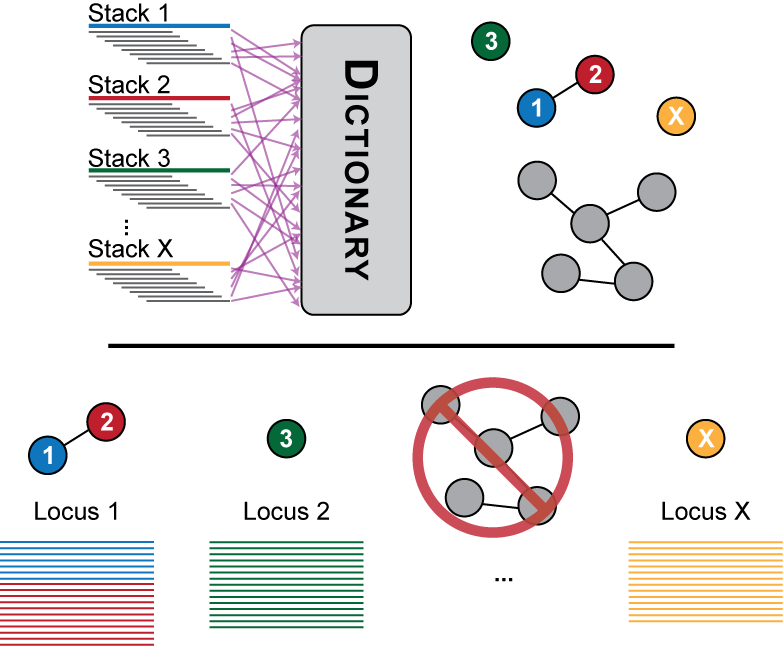
Figure 2. The consensus sequence from each stack is broken down into kmers in the second stage of the ustacks algorithm and stored in a dictionary. Two stacks that have a certain number of k-mers in common are considered as potentially matching and are aligned together. If the number of nucleotide mismatches are less than the distance allwoed between stacks the stacks are merged into a locus.
Once a set of exactly matching stacks has been generated, the second stage of the algorithm seeks to match putative alleles together into a locus. The distance allowed between stacks parameter represents the number of nucleotides that may be different between two stacks in order to merge them. These nucleotide differences may be due to polymorphisms present between two alleles, or they may be due to sequencing error.
In Figure 2, Stack 1 and Stack 2 are found to have fewer nucleotide mismatches than allowed by the distance parameter and are merged into polymorphic Locus 1. Stack 3 and Stack X are found to be monomorphic and are converted to individual Locus 2 and Locus X. The large grey set of stacks represents a set of repetitive sequence that has too many alleles to be biologically correct. The pipeline detects these loci and blacklists them from the rest of the pipeline.
Once the loci are formed, the secondary reads are brought back into the analysis and are aligned against the assembled loci using a more permissive nucleotide mismatch value (you can control this value with the -N parameter to ustacks or denovo_map.pl). This process provides more depth which aides the SNP calling model in detecting polymorphisms. A locus with a single polymorphism is outlined in Figure 3.
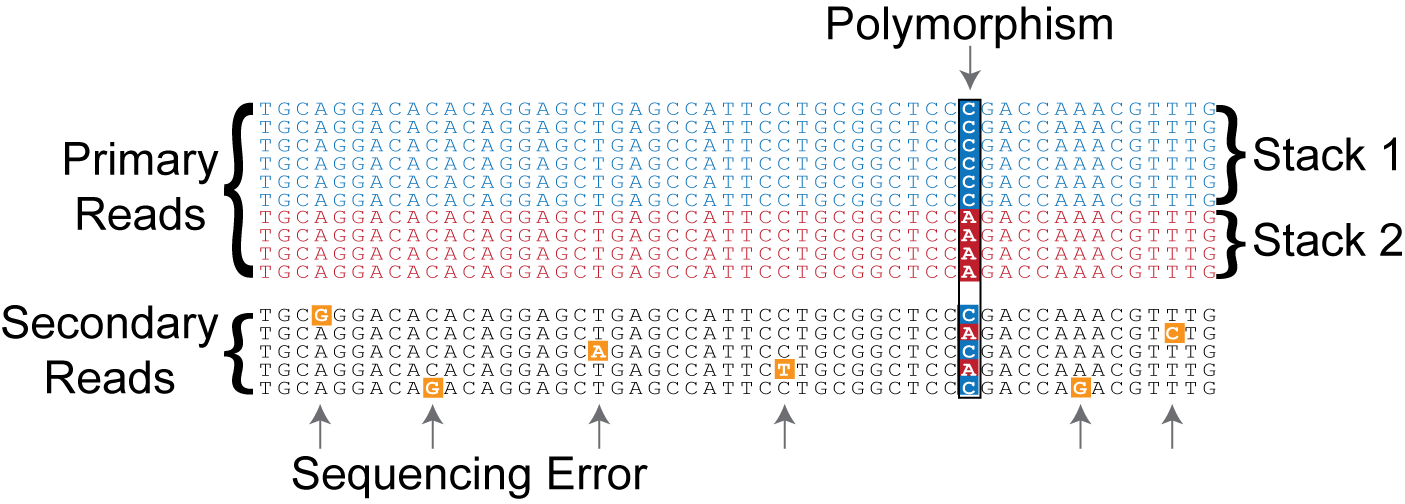
Figure 3. The major components in a Stacks Locus.
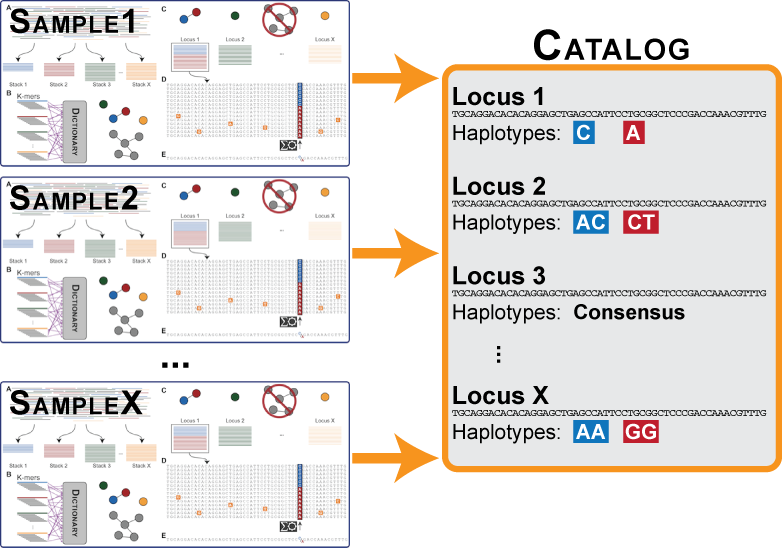
Figure 4. Catalog construction.
The ustacks program will be executed on each individual sample in the data set to build loci. Once this is complete, the data from each individual will be merged into a catalog (by the cstacks program), which is meant to contain all the loci and alleles in the population. In the case of a mapping cross, then the catalog can be built soley from the parental loci. In the case of a population, the catalog will be constructed from the loci in each individual in the population.
Rarely in the case of a mapping cross, but frequently in the case of a population, there will be monomorphic, or fixed loci in two or more individuals in the population. However, if you compare these loci to one another, you will find that the are differentially fixed versions of the same locus and should be merged into a single locus in the catalog. If the distance between catalog loci parameter is greater than 0, then cstacks will use the consensus sequence from each locus to attempt to merge loci together across samples.