How do genomes change over evolutionary time? Numerous questions fall under this rubric, such as:
These questions can be addressed at several different scales, from whole genome duplication over millions of years to structural variation within a species over thousands of years, to somatic evolution and cancer within an individual over a handful of years. The research strategy of the Catchen Lab is to apply novel algorithmic developments to the questions of genome evolution.
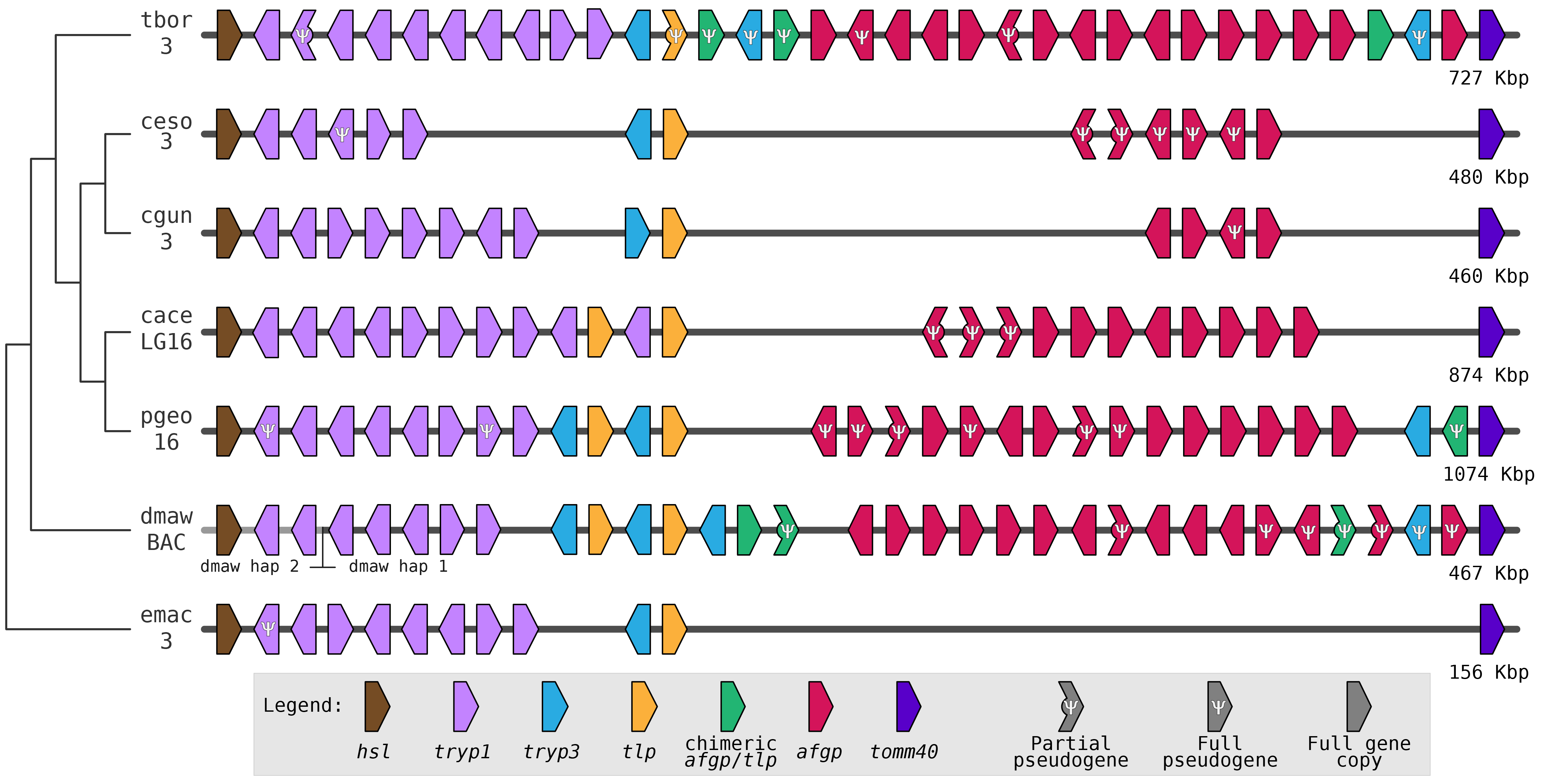
The lab's work is mostly focused on teleost fish. Teleosts represent the most species rich vertebrate clade and within the teleosts lie laboratory model organisms, such as zebrafish, and several fascinating natural evolutionary models, including the threespine stickleback. We have projects focused on a number of species, from Antarctic notothenioids, to stickleback, killifish, and several modern and ancient salmon species.
 |
Jack Calvery, Graduate Student |
 |
Julian Catchen, Principal Investigator |
 |
Akane Hatsuda, Postdoctoral Research Associate |
 |
Gio Madrigal, Graduate Student |
 |
Bushra Minhas, Graduate Student |
| ⇒ Former lab members |
|
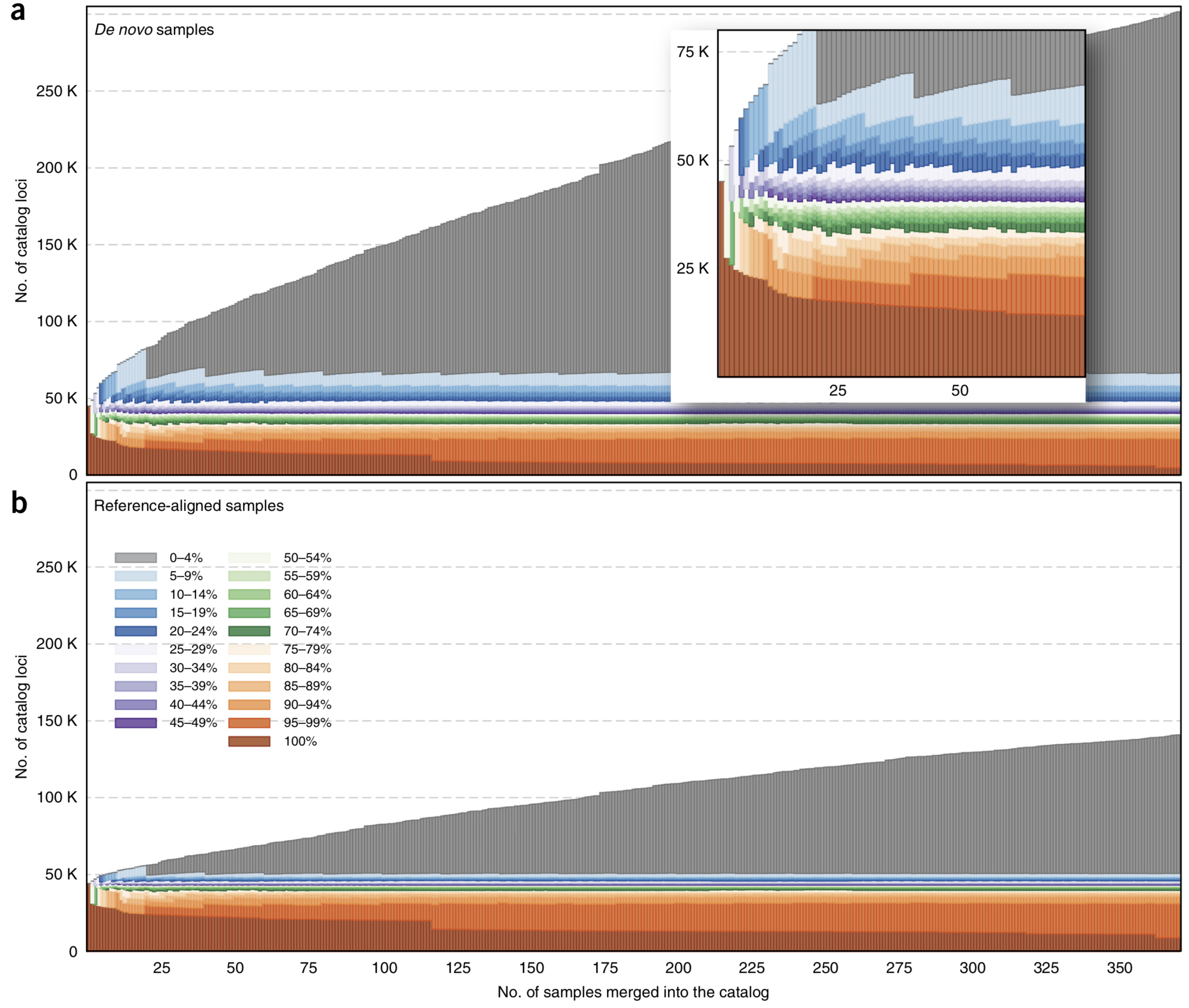
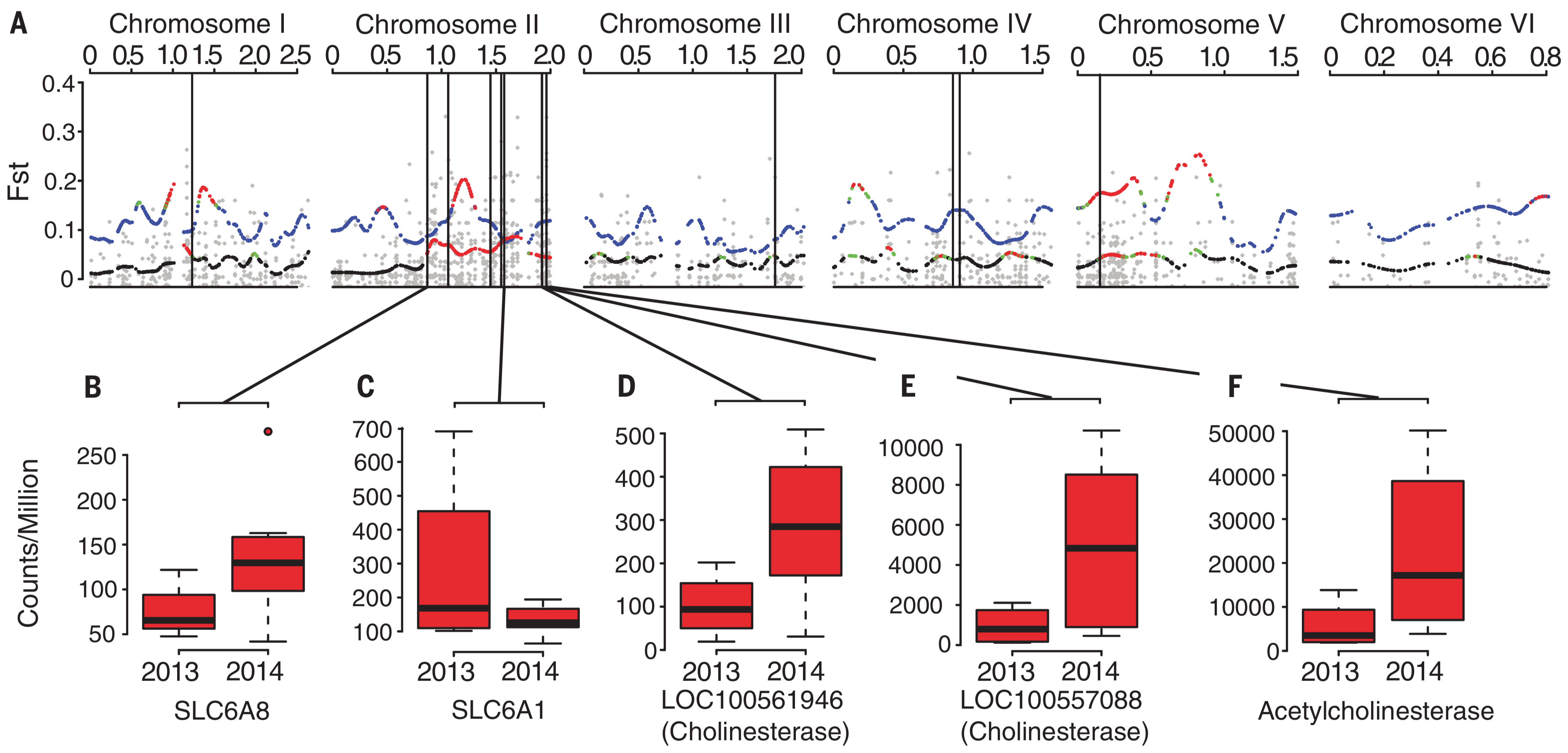
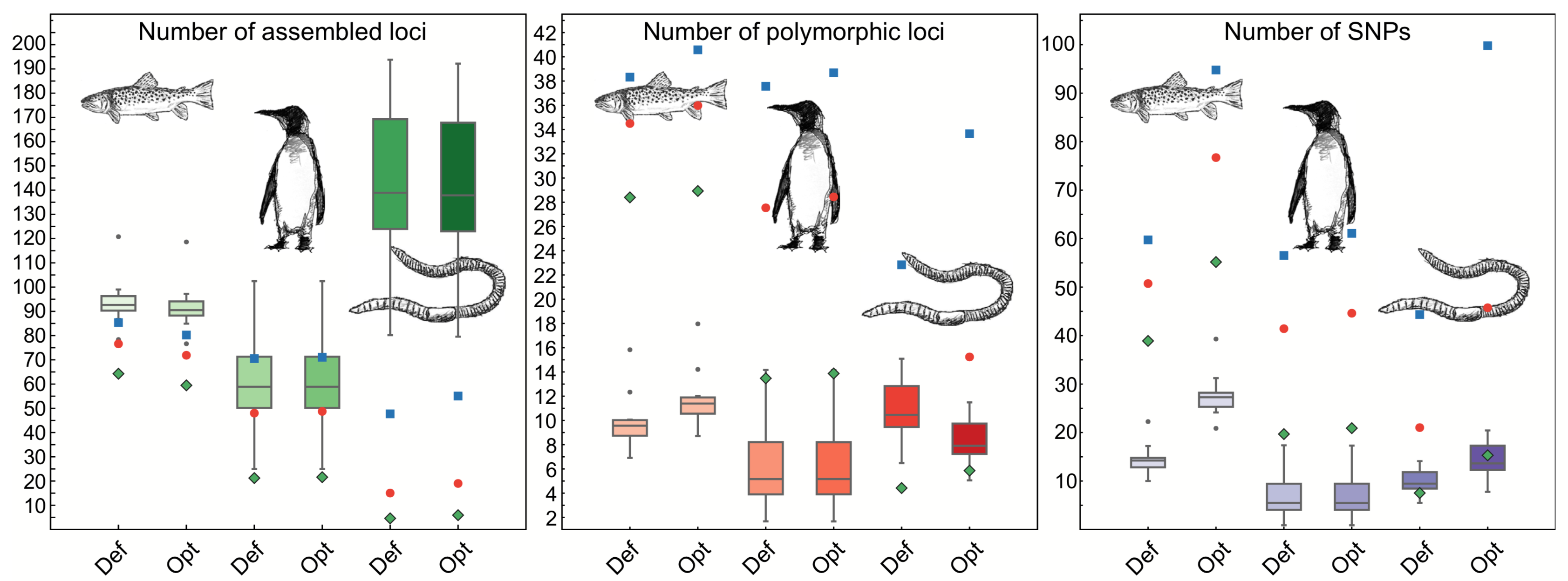
Stacks is a parallelized software system that can assemble and genotype tens of thousands of restriction enzyme-based markers in thousands of individuals. Stacks can be used to develop ultra dense genetic maps, or it can be used to identify evolutionarily divergent segments of the genome using population genomic statistics such as π and FST. |
|
|
Klumpy is a tool designed to look for either missassembled regions in a long-read genome assembly or a set of query sequences in another set of sequences (e.g., finding genes of interest in a reference genome). Results can be visualized alongside other features (e.g., sequence gaps) for easier interpretation. |
|
|
RADinitio is a forward simulator for creating population-level RAD data sets, based on a given reference genome. This in silico RADseq library preparation and sequencing process, allows for the exploration of parameters including restriction enzyme selection, library insert size, PCR duplicate distribution, and sequencing coverage. |
|
|
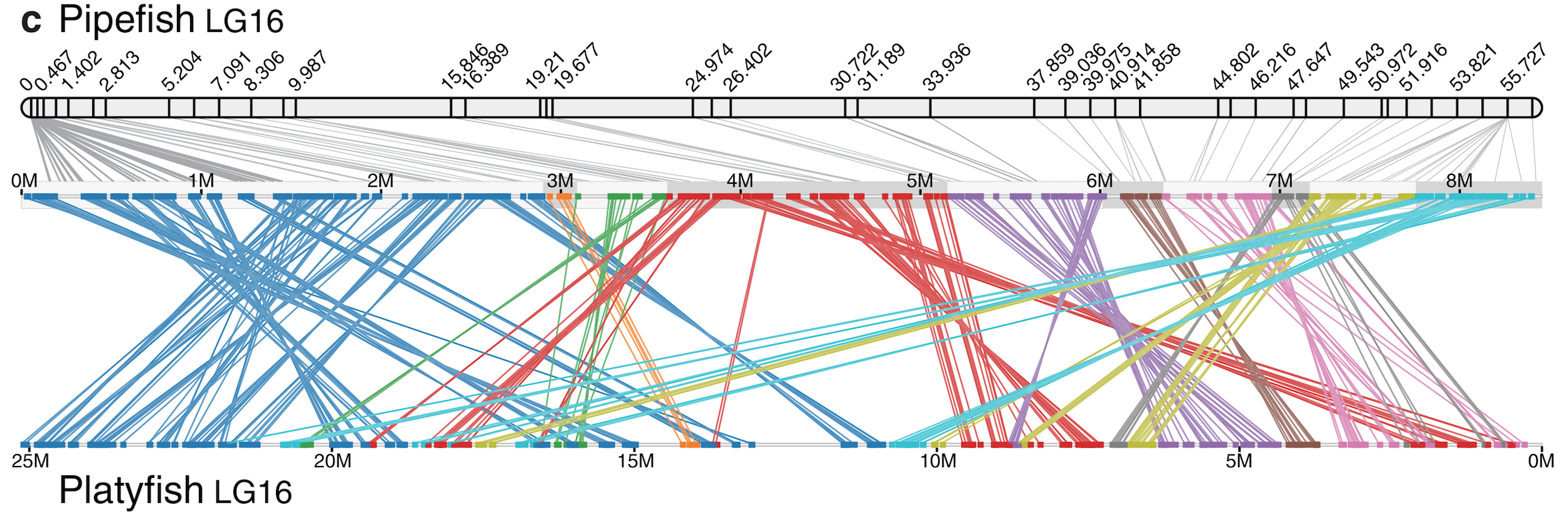
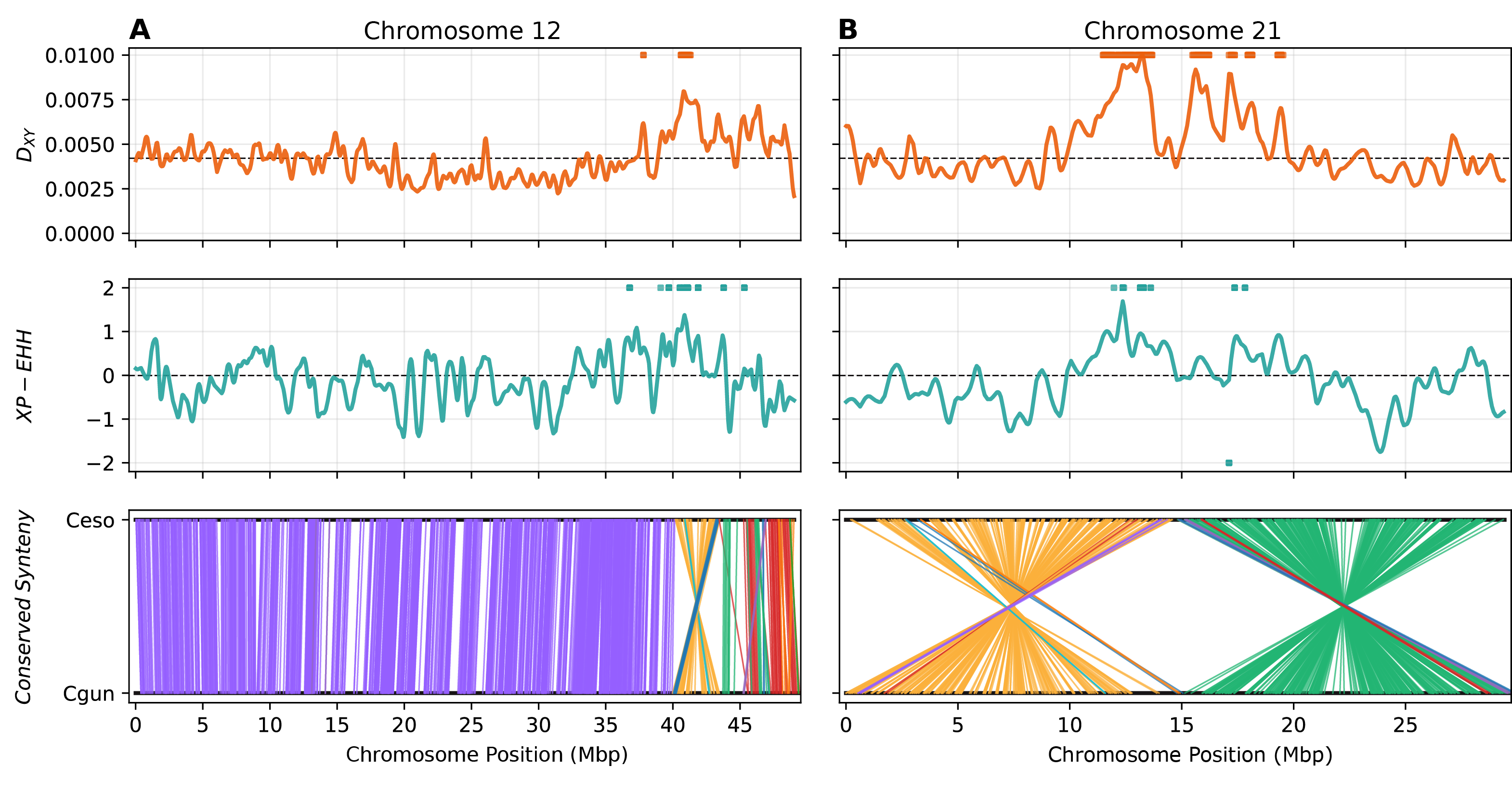
Chromonomer is a program designed to integrate a genome assembly with a genetic map. |