Julian Catchen1, Angel Amores2
|
1Department of Evolution, Ecology, and Behavior University of Illinois at Urbana-Champaign Urbana, Illinois, 61820 USA |
2Institute of Neuroscience University of Oregon Eugene, Oregon, 97403-1254 USA |
Chromonomer is a program designed to integrate a genome assembly with a genetic map. Chromonomer tries very hard to identify and remove markers that are out of order in the genetic map, when considered against their local assembly order; and to identify scaffolds that have been incorrectly assembled according to the genetic map, and split those scaffolds.
Chromonomer uses the standard autotools install:
% tar xfvz chromonomer_x.xx.tar.gz % cd chromonomer_x.xx % ./configure % make (become root) # make install
You can change the root of the install location (/usr/local/ on most operating systems) by specifying the --prefix command line option to the configure script.
% ./configure --prefix=/home/smith/local
A successful build will place the chromonomer program in /usr/local/bin and create a directory (by default) in /usr/local/share/chromonomer to hold the web interface.
Add the following lines to your Apache configuration to make the Chromonomer PHP files visible to the web server and to provide a easily readable URL to access them:
<Directory "/usr/local/share/chromonomer/php"> Order deny,allow Deny from all Allow from all Require all granted </Directory> Alias /chromonomer "/usr/local/share/chromonomer/php"
A sensible way to do this is to create the file chromonomer.conf with the above lines.
Place the chromonomer.conf file in
/etc/apache2/conf-available
directory. Then restart Apache. Like so:
# vi /etc/apache2/conf-available/chromonomer.conf # ln -s /etc/apache2/conf-available/chromonomer.conf /etc/apache2/conf-enabled/chromonomer.conf # apachectl restart
First you must align your markers against the newly built reference genome. Be very careful about allowing too much promiscuity in your alignments as this will lead to spurious marker alignments that will cause more scaffolds to be split. Restrict the usage of gaps in the alignment to the best extent possible.
To do the alignments, one method is to create a FASTA file containing the sequence of each marker. The ID of each marker must match the ID of the markers that you provide to describe your genetic map. If you used Stacks to generate your markers, it is easy to export the consensus sequence for each marker from the Stacks Catalog using a few UNIX commands. Here is an example of a FASTA file containing markers, which can be fed to an alignmnet program (such as BWA or GSnap):
>10311 TGCAGGTCATCAAACCTGCCTCCACACTGGTGAGCTCAAGAAATTCCCACAAATGTTGTTGTCCCCAAAAAACTTCTTTTTTTTGTTTGGGAGTT >1825 TGCAGGCGACTCACGCGGTCCTCACGGGCACCCTGGTGCCCGCGGGCATCGTGTTGGTGACCCCTGCTGTAGAAGGTACCTAAATGCACCACAGC >19504 TGCAGGAAGTTCAGCGAGCGCGTTCAGCGAGCGCGTTCAGCGAGCGCGTATAGCGAGGTGTGACTCCAACGACGATATTAATGAGCTTTGGGATA >11977 TGCAGGACGGAGGTGCTCATACAGACATGTATCGACTTCAAATCAGTACTCGTTTTTTTAATGCGCGGAAAAGCAAGTTGCGCGACATTTTACGC >16603 TGCAGGATGTGTTTGGAGCACATTGTGAGATTCAAACTTTCAAAACAAAGAAACTAGCGTCTCCCACTACATGTACCTTTATGTACTCTATCCAG >10785 TGCAGGAAATTGAGAAAGAGAACAAGAACTCCCAGCATAAACCAGGTGAGAATTGTCATCCTTGGGAATGTTCACGAGATTTACACAATCTCTGC ...
The result of the alignment should be a SAM or BAM file, which will contain the marker IDs and the alignment positions of each marker.
You will need a tab separated list describing your genetic map -- the markers in the map along with their linkage group and centiMorgan position. Chromonomer was designed to handle genetic maps built from RAD data using Stacks ( http://catchenlab.life.illinois.edu/stacks) although any genetic map where the markers can be aligned to the genome should work. The file should be formated in the following way:
Linkage Group<tab>Locus (Marker ID)<tab>cM Position
Here is an example:
lg1 10311 0 lg1 1825 0 lg1 19504 0.958 lg1 11977 0.97 lg1 16603 0.97 lg1 10785 0.97 lg1 18192 0.97 lg1 16462 0.981 lg1 17763 0.981 ... lg1 585 64.061 lg1 11929 64.332 lg2 6504 0 lg2 2294 0 lg2 13138 0.785 lg2 11849 1.887 lg2 18900 1.887 ...
Create a directory to hold the Chromonomer output. For example, 20150603. Create a directory with the same name under the web interface:
% mkdir /usr/local/share/chromonomer/php/20150603
Run Chromonomer, specifying the proper input and output paths and providing the directory you created using the --data_version flag. For example:
% chromonomer -p ~/research/20150603_linkage_map.tsv \ -o ~/research/20150603/ -s ~/research/markers.sam \ -a ~/research/final.assembly.agp --data_version 20150603
For Chromonomer to work, it must be able to sync the markers in the genetic map with the markers in the SAM/BAM alignments, and it must be able to sync the scaffold names in the AGP file with the scaffold names in the SAM/BAM alignment files. Depending on where you got your AGP file and your genetic map description, you may need to edit these files to make sure all the IDs match (e.g. sometimes the scaffold IDs in the AGP file have additional verbiage not present in the FASTA file that contained your genome assembly).
Once Chromonomer is complete, you can copy the files from the output directory to the web interface:
% sudo cp ~/research/20150603/* /usr/local/share/chromonomer/php/20150603/.
You should be ready to view the output in the web interace.
As you make corrections to your map, you will re-execute chromonomer. It is easy to create another directory under the web interface and store the results there. This way you can keep versions of the assembly around for comparison as you make imrpovements.
Chromonomer creates a voluminous output, trying to document how and why the genome was (or was not) assembled. A number of files are precomuted for the web interface, but almost all of the same inforamtion is available in the text output.
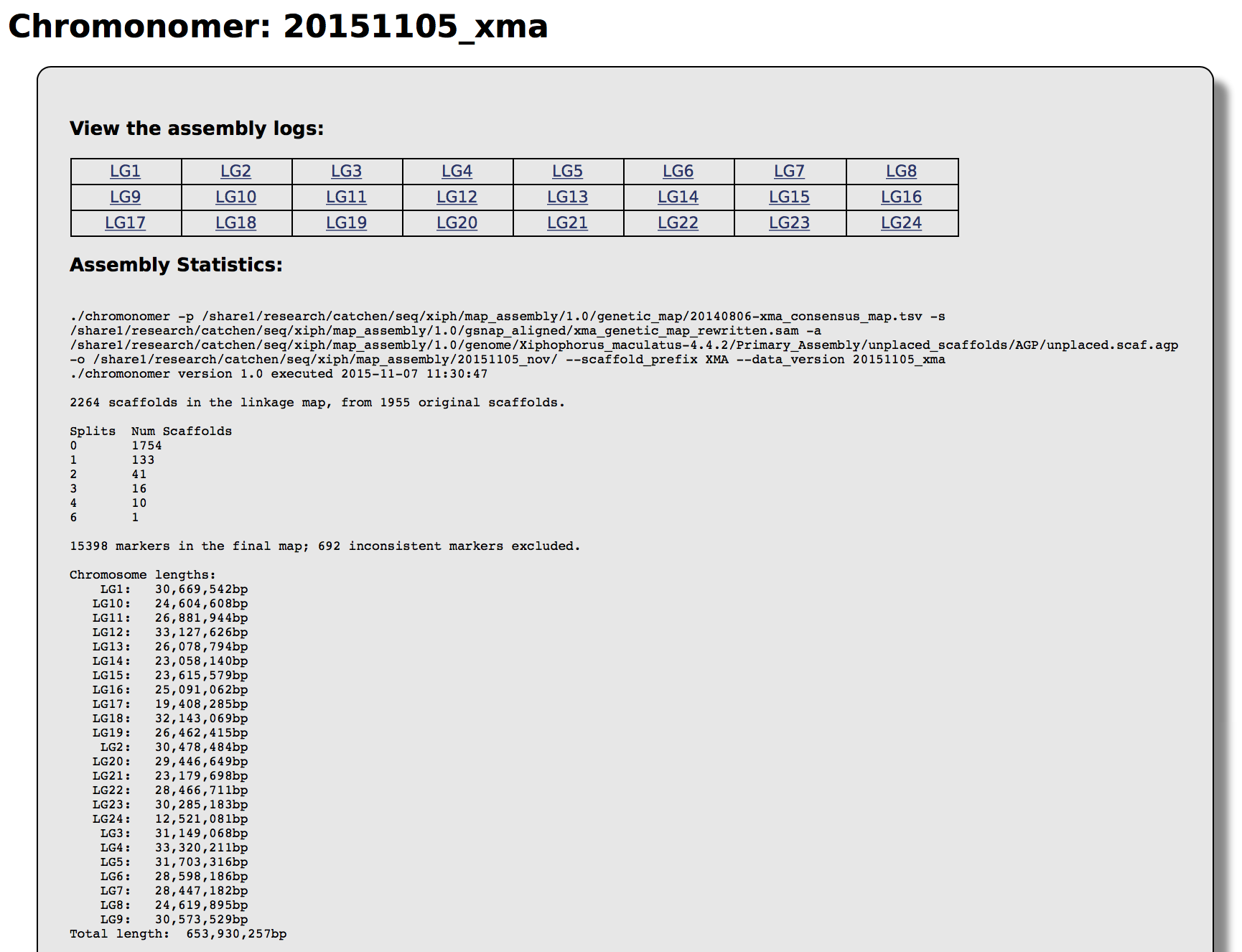
| chromonomer_summary.log | Records the date/time Chromonomer was run along with the exact command. The file contains a summary of the run, including number of scaffold splits and the total length of each chromosome. |
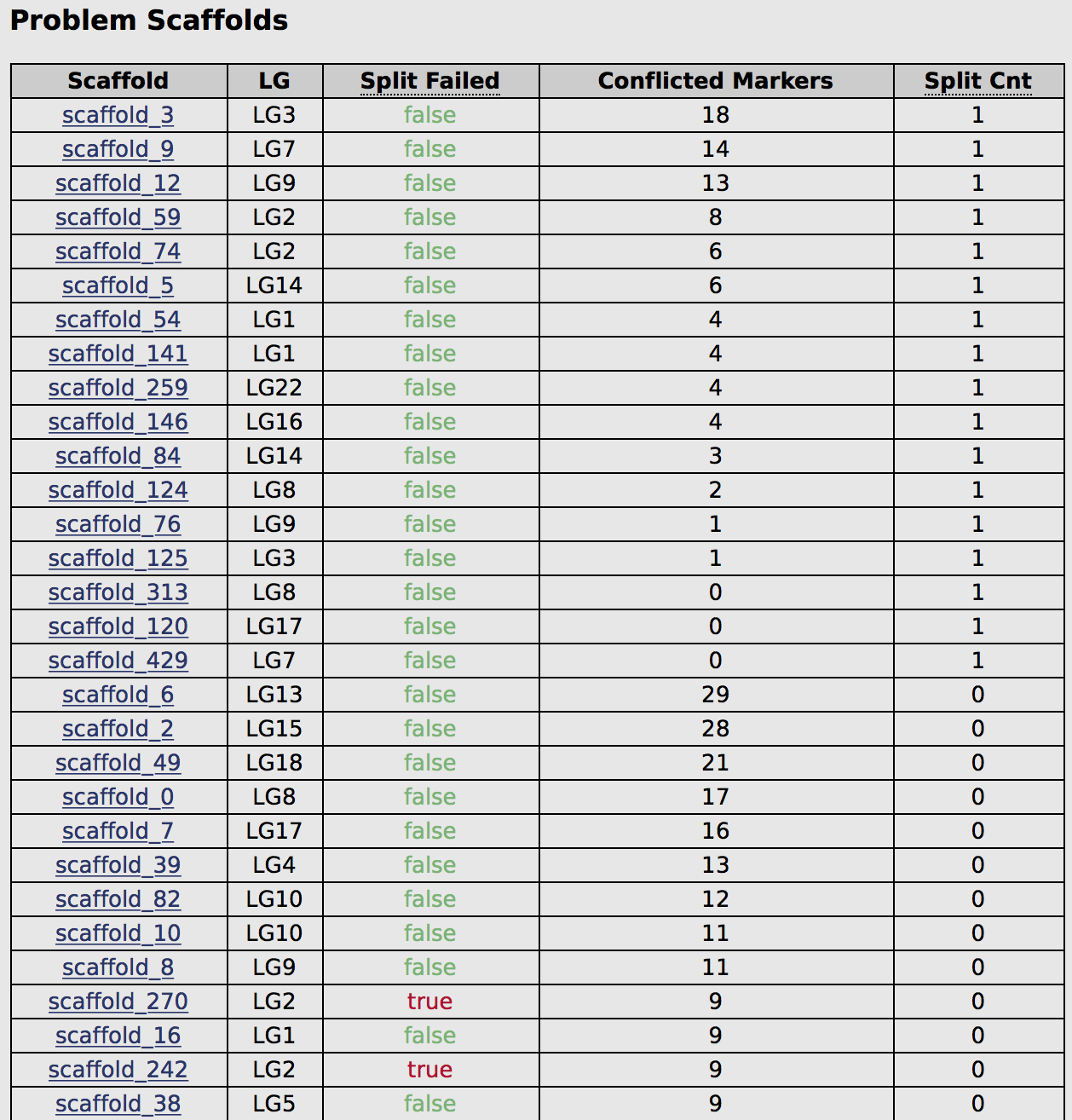
| problem_scaffolds.tsv | Contains a list of all scaffolds where a split occurred, where a split was attempted but failed, or where markers were pruned. The file is printed in a sorted order to help prioritize which scaffolds were the most problematic. |
| split_scaffold_map.tsv | Contains a list of all scaffolds that were split and the coordinates upon which they were split. |
| XXX_genome.agp | An AGP file that contains a definition of the final, chromonomed assembly. Each linkage group, with its complement of scaffolds and gaps is defined. Scaffolds are defined in terms of their original names, with basepair boundaries of each scaffold modified to represent any scaffold splits. |
| XXX_unplaced_scaffolds.agp | Scaffolds that could not be integrated into the genetic map are placed in this file, unmodified from their original input. |
| XXX_scaffolds.agp | An AGP file that remains defined at the scaffold level (no chromosomes are present) with the scaffold splits defined. The scaffolds generated by splitting are listed here, with their constiuent contig/gap components along side the other scaffold components that were not split. |
| scaffold_XXX.log | A record of what happened to each scaffold that was in any way modified (with markers pruned or split). See the next section for details as to how interpret these files. |
|
LGX_before.php LGX_after.php LGX_before.json LGX_after.json |
These files are used by the web interface to report what a linkage group and set of scaffolds looked like before Chromonomer integrated them and after the integration is complete. The *.json files feed the web visualizations of each linkage group. |
The web interface places all of the information from a Chromonomer integration into a single place and links pieces of the integration together, allowing for easy access. You can easily place successive runs of the software together and the web interface will allow you to compare them:

For a particular integration, the web interace will give you access to all the linkage group information along with the summary statistics from the integration (these are also available in the chromonomer_summary.log file):
The web interace will also give you fast access to all the problem scaffolds, in a usefully sorted order (these are also available in the problem_scaffolds.tsv file):
Finally, for each linkage group, the web interface will draw a before and after visualization, so you can see how the scaffolds go together, where the markers fall, and providing links to the scaffold logs for each modified scaffold:
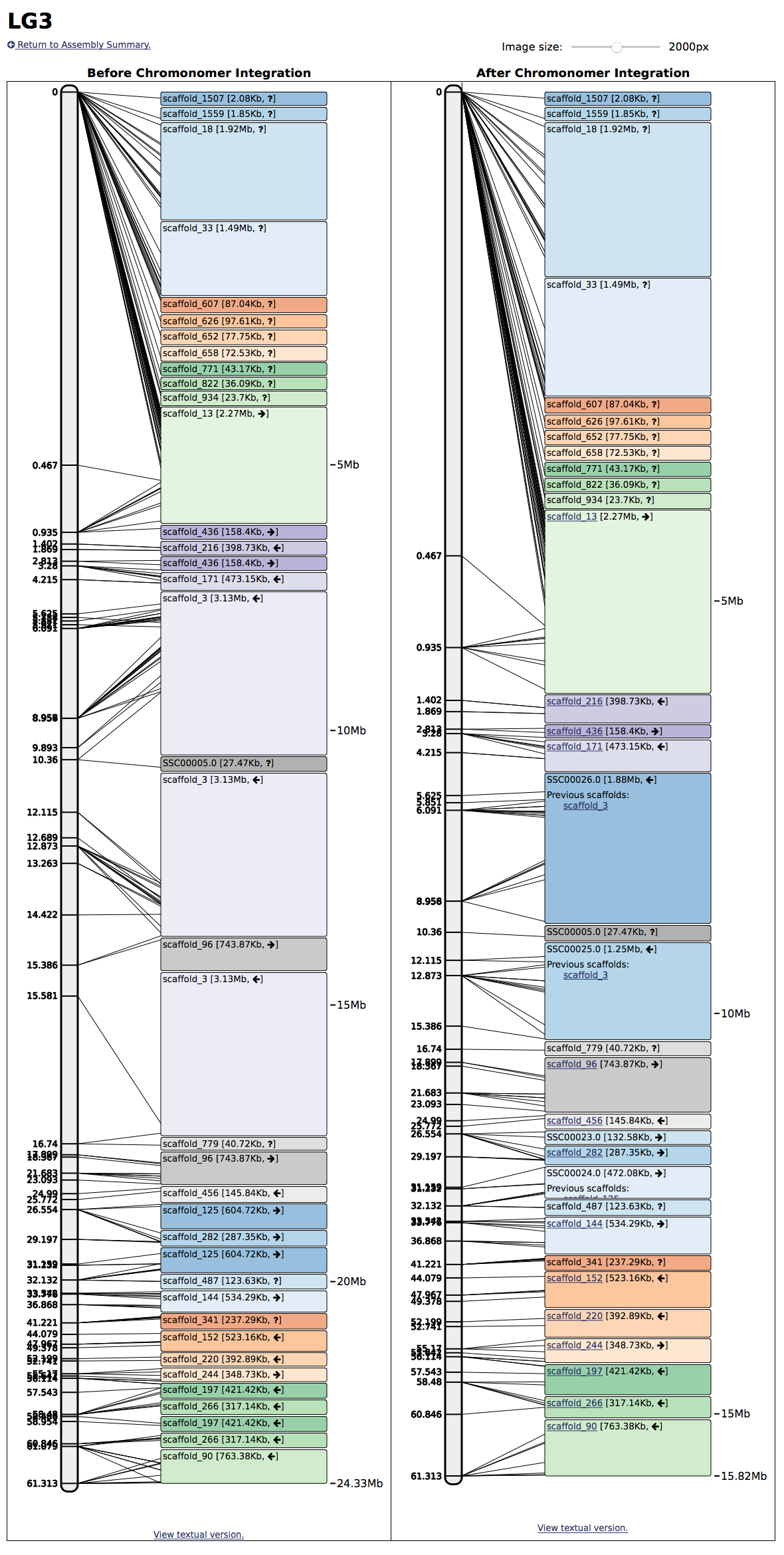
Chromonomer tries to document all of the sources of data available to it while it is integrating scaffolds with the genetic map. For each scaffold, a log file is kept to record any conflicting markers found, or any splits that were made to the scaffold. These scaffold logs can be used to identify markers with a large amount of genotyping errors or to identify scaffolds that were likely assemnbled incorrectly.
Once data is read into memory, the first thing Chromonomer will do is to check that each scaffold is anchored to only a single linkage group. Since it would require a large amount of error for markers to be assigned to the wrong linkage group by the linkage mapping program, Chromonomer assumes that the map assignment is correct and will split any scaffold assigend to two or more linkage groups.
Chromonomer will check that each scaffold is mapped to a single, continuous part of the linkage group. If the scaffold spans more than one node in the linkage group, then its orientation can be determined.
If a scaffold exists on two subsets of map nodes that are not continuous, then there are other scaffolds in the assembly that fall in the between the current scaffold. This can be caused either because of an assembly error, e.g. the scaffolding algorithm made an incorrect join, of because there is another scaffold that occurs inside a gap of the existing scaffold.