Chromonomer is a program designed to integrate a genome assembly with a genetic map. Chromonomer tries very hard to identify and remove markers that are out of order in the genetic map, when considered against their local assembly order; and to identify scaffolds that have been incorrectly assembled according to the genetic map, and split those scaffolds.
Given a discription of an assembly, and a description of a genetic map, Chromonomer will try to reconcile them. Chromonomer accepts standard files to perform this analysis.
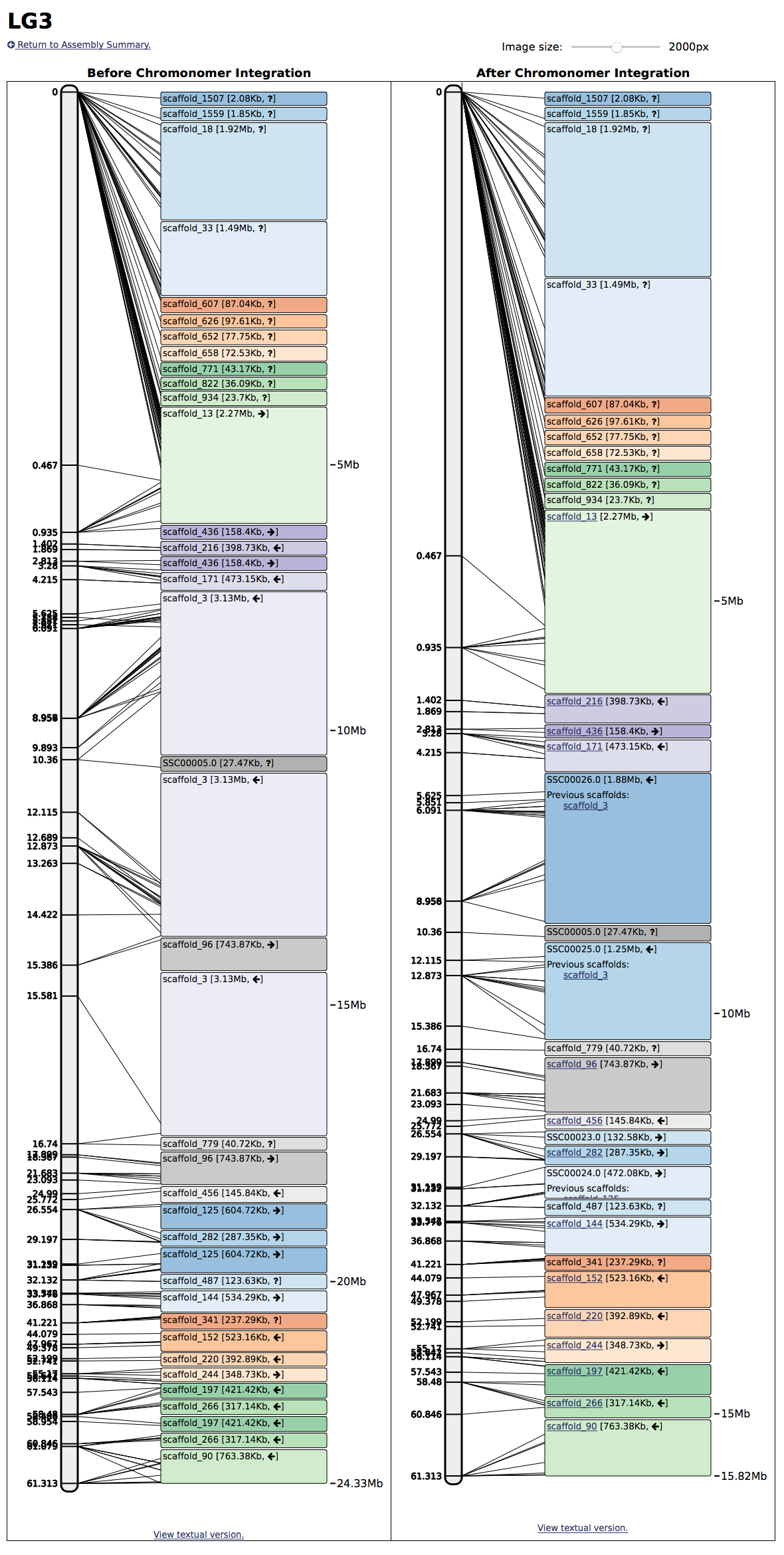
A visualization of Chromonomer integrating a genetic map and genome assembly (left side, before, right side, after).
Depending on the number of progeny in the genetic map (which limits the number of recombination events), and the level of genotyping error, a genetic map will contain a certain amount of uncertainty and error in how the markers are ordered. Similarly, depending on how a genome assembly was scaffolded, there can be incorrect joins in the scaffolds. Chromonomer provides the best chromonome-level assembly (a.k.a. chromonome) given the amount of information in supplied data.
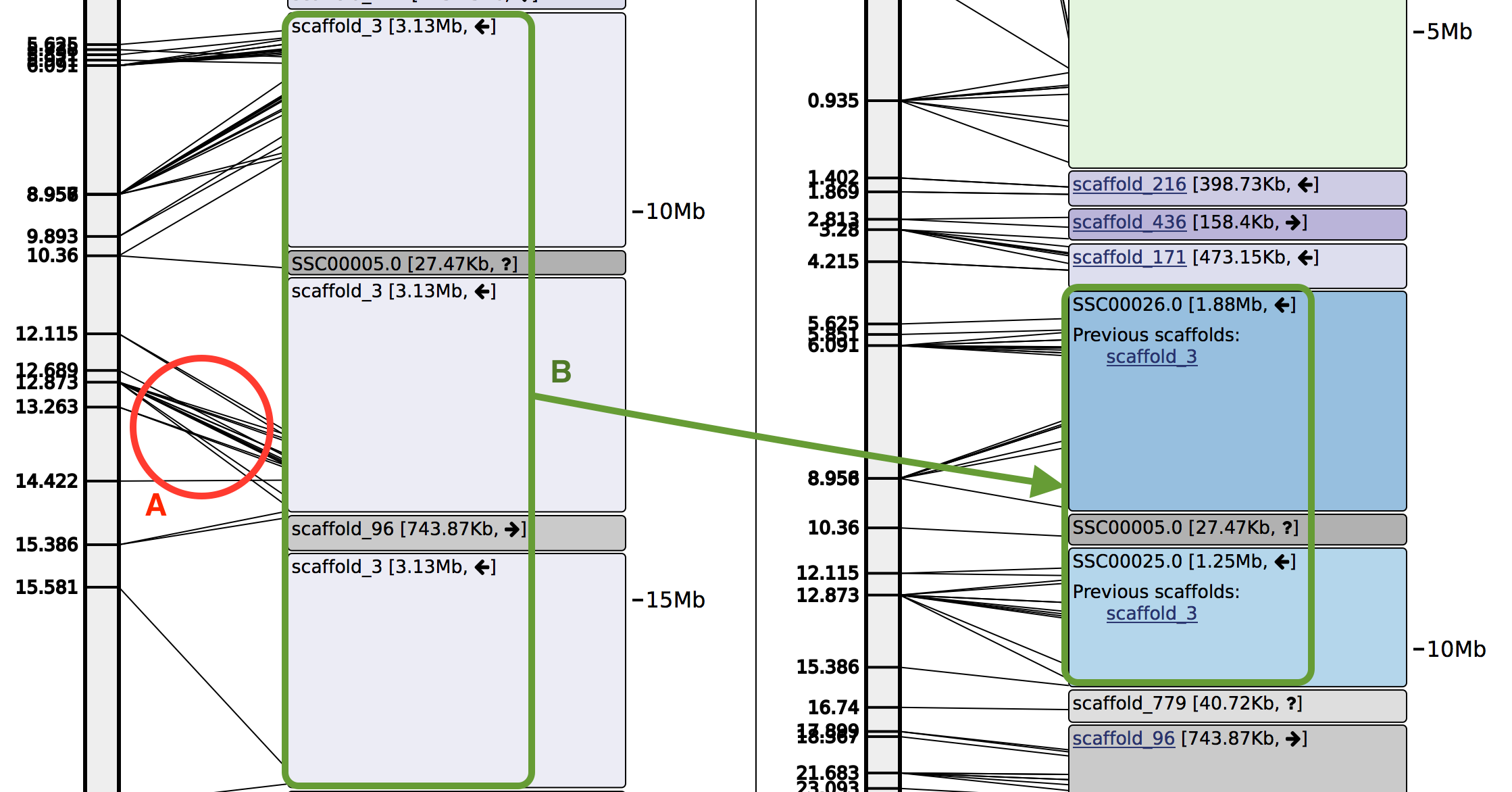
Two examples of modifications Chromonomer will make. A. Markers (displayed as lines connecting the linkage group to the ordered scaffolds) are in order with respect to the map, but out of order with respect to the local assembly area. Chromonomer will prune the minimal number of markers to get a consistent ordering. B. Scaffold_3 can be seen in three places on the linkage group, with distinct sets of markers (left side). Chromonomer places scaffold_3 in a single location in the map, then splits it (into scaffolds SSC00025 and SSC00026) to accommodate scaffold_779 which is gap-filled in between the two.
Chromonomer is implemented in C++. It will compile on GNU-based Linux systems or BSD-based (Apple) OS X systems. Chromonomer is released under the GNU GPL license.
Chromonomer was developed by Julian Catchen <> with contributions from Angel Amores <>.
The Chromonomer preprint describing the software is now available on bioRxiv:
Chromonomer has been used to chromonome several genomes, including these two: